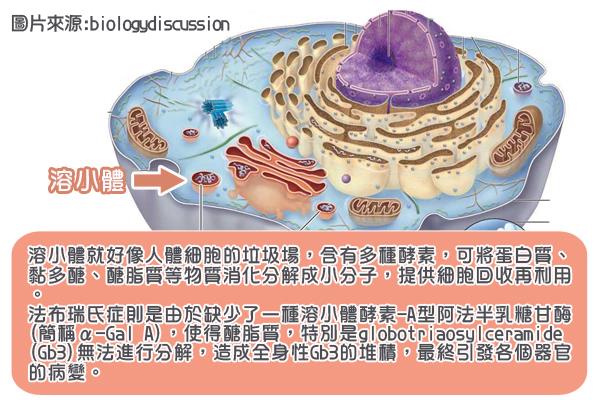
法布瑞氏症是一種罕見疾病,為溶小體儲積症的一種。何謂溶小體? 溶小體如同人體細胞的資源回收場,在正常狀況下,溶小體含有多種酵素,可將蛋白質、黏多醣、醣脂質等物質消化分解成小分子,提供細胞回收再利用。法布瑞氏症因為缺少了一種溶小體酵素—A型阿法半乳糖酐酶(α- galactosidase A;簡稱α-Gal A),使得醣脂質,特別是globotriaosylceramide(簡稱Gb3)無法進行分解,於是堆積在全身許多細胞的溶小體內。
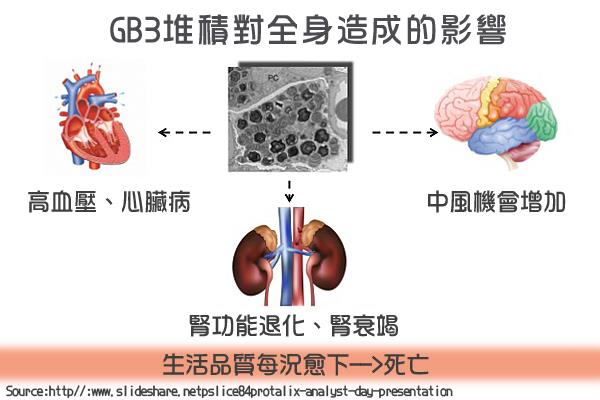
過多的Gb3在體內會到處堆積,主要堆積器官有心臟、腎臟,包括:腎絲球與腎小管的上皮細胞;心肌細胞與瓣膜纖維細胞;背根神經節的神經元與自主神經系統;角膜上皮細胞;血管內皮、外皮、平滑肌細胞,最終引發各個器官的病變。
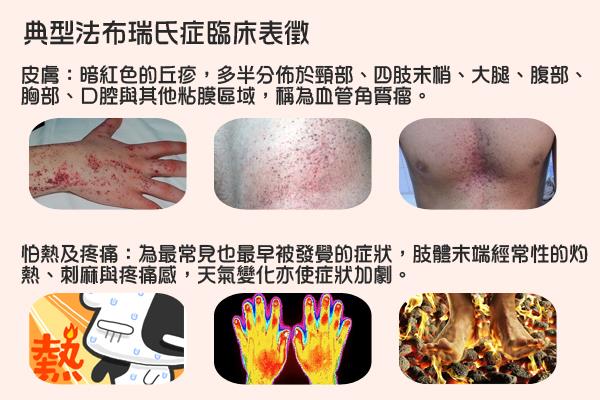
法布瑞氏症的病患,兒童與青少年期開始出現臨床症狀,典型的法布瑞氏患者會出現肢體末端間歇性的疼痛、燒灼痛感,許多皮表會呈現暗紅色斑點,包括頸部、四肢末梢、大腿、腹部、胸部等。到了成年之後,出現進行性的腎臟、心血管及腦血管病變,成為威脅生命的主因。
這種疼痛伴隨燒灼般的特性,當肢體疼痛來襲時,有些患者甚至痛到掉淚、無法站立而嚴重影響生活品質,患者甚至說老師都以為他們是偷懶不想上課,飽受異樣眼光看待,甚至面臨休學、放棄工作的窘境。此病在早期很難被診斷出來,從病患發病開始平均大約需要10到20年的時間才能確立診斷。
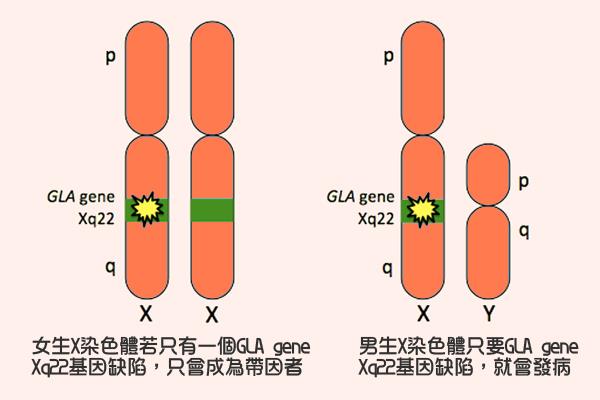
法布瑞氏症的缺陷基因GLA gene位於X染色體,屬於性聯遺傳疾病,因此對男性的影響會較大,男性只要有一個X染色體有該基因缺陷便會發病,女性帶因者(一個X染色體為正常,一個有基因突變)也會出現病狀,但通常較為輕微。台灣在2006年將Fabry disease列為新生兒篩檢項目之一,透過篩檢,測定寶寶血液中α-Gal A酵素活性,及早診斷法布瑞氏症。
法布瑞氏症還有另一個型態—心臟變異型,正是Fabry disease裡比較不容易被早期診斷的變異型,因為心臟變異型的患者,其體內的A型阿法半乳糖酐酶尚有部分活性,這些殘存的酵素活性,會幫助脂質代謝,絕大部分這些病人的問題特別出現在心臟方面,而且患者在幼年和青少年時期症狀不明顯,通常要到40-50 歲左右問題才會浮現。
法布瑞氏症在過去僅能採取症狀治療,隨著基因工程技術的進步,使酵素替代療法成功的運用在治療上。它不僅止於改善症狀,更可進一步延長壽命與提高生活品質。患者若能及早診斷,並依照醫師指示接受治療並定期回診追蹤,此罕見疾病並非絕望的不治之症,及早治療亦可擁有一般無二的大眾生活。